

El síndrome de Vogt-Koyanagi-Harada (VKH), también conocido como síndrome uveomeníngeo, es una panuveítis granulomatosa bilateral y difusa que cursa con desprendimiento de retina seroso y que puede acompañarse de afectación del sistema nervioso central, alteraciones dermatológicas y auditivas.
Su nombre deriva de los autores que la describieron por primera vez. Afecta a adultos de ambos géneros, entre los 20 y 50 años de edad, y presenta una prevalencia aumentada en razas negras. Este síndrome inflamatorio probablemente sea el resultado de un mecanismo autoinmune, influenciado por factores genéticos.
La evolución de la enfermedad se divide en 4 estadios clínicos: prodrómico, uveítico agudo, de convalecencia y crónico recurrente. El diagnóstico es fundamentalmente clínico, mediante los criterios establecidos por la Sociedad Americana de Uveítis (AUS) publicados en el año 2001. Es necesario realizar diagnóstico diferencial con la oftalmía simpática, el linfoma primario de células B, la escleritis posterior y el síndrome de efusión uveal. El tratamiento precoz y mantenido es la base de una buena evolución.
La enfermedad se presenta en 4 fases distintas: Fase prodrómica: Suele durar desde días a semanas, generalmente una semana. Se presenta en forma de clínica seudogripal con fiebre, cefalea, náuseas y dolor orbitario. Suele cursar con tinnitus y, menos frecuentemente, con manifestaciones neurológicas. Fase uveítica aguda: Se inicia unos días después de la fase prodrómica y puede durar entre 2 y 3 meses. Se caracteriza por una uveitis bilateral, aunque en algunos casos puede afectarse primero un ojo y posteriormente bilateralizarse, con la apariención súbita de visión borrosa, fotofobia y dolor ocular. Puede acompañarse de disacusia y meningismo. Fase crónica o convalecencia: Aparece a los 3 meses de la fase aguda. En ella son más frecuentes los síntomas cutáneos, aunque estos pueden aparecer antes, durante o después del compromiso ocular. Se han descrito canicie, poliosis, alopecia y vitíligo, que en algunas ocasiones se presenta siguiendo una distribución neural, acompañándose de parestesias y disestesias. Afecta frecuentemente a la cara, el cuello, el tronco y los párpados. Fase crónica recurrente: Tiene lugar a los meses-años después de la fase aguda, interrumpiendo la fase de convalecencia. Esta fase no está presente en todos los pacientes. Se caracteriza por episodios de uveítis anterior granulomatosa frecuentemente resistente a tratamiento esteroideo. De forma infrecuente se asocia a uveítis posterior. Característicamente suelen aparecer nódulos de iris.
Se describen 3 tipos de síndrome de VKH: Tipo I: hay compromiso ocular sin compromiso auditivo ni cutáneo, que permite sospechar el VKH.
Tipo II: hallazgos oculares, con al menos una manifestación auditiva o cutánea. Tipo III: el paciente presenta afectación oftalmológica, auditiva y cutánea.
Aproximadamente el 70% de los casos del tipo I y II tienen una duración de la enfermedad de menos de un año, mientras que dos tercios de los casos del tipo III presentan enfermedad activa por más de un año. No hay relación entre la gravedad del compromiso visual y la de la enfermedad general, pero los pacientes con afectación tipo III tienen curiosamente mejor agudeza visual final que aquellos de tipo I y II.
En cuanto al tratamiento no existe una pauta establecida a seguir. Se han utilizado diferentes fármacos en el tratamiento del VKH recidivante, con eficacia variable. Los glucocorticoides sistémicos son el fármaco de elección en los brotes agudos. La dosis inicial, la duración del tratamiento y la pauta descendente deben individualizarse en cada paciente. También, se han empleado varios fármacos como los inmunosupresores, los fármacos biológicos y las inmunoglobulinas.
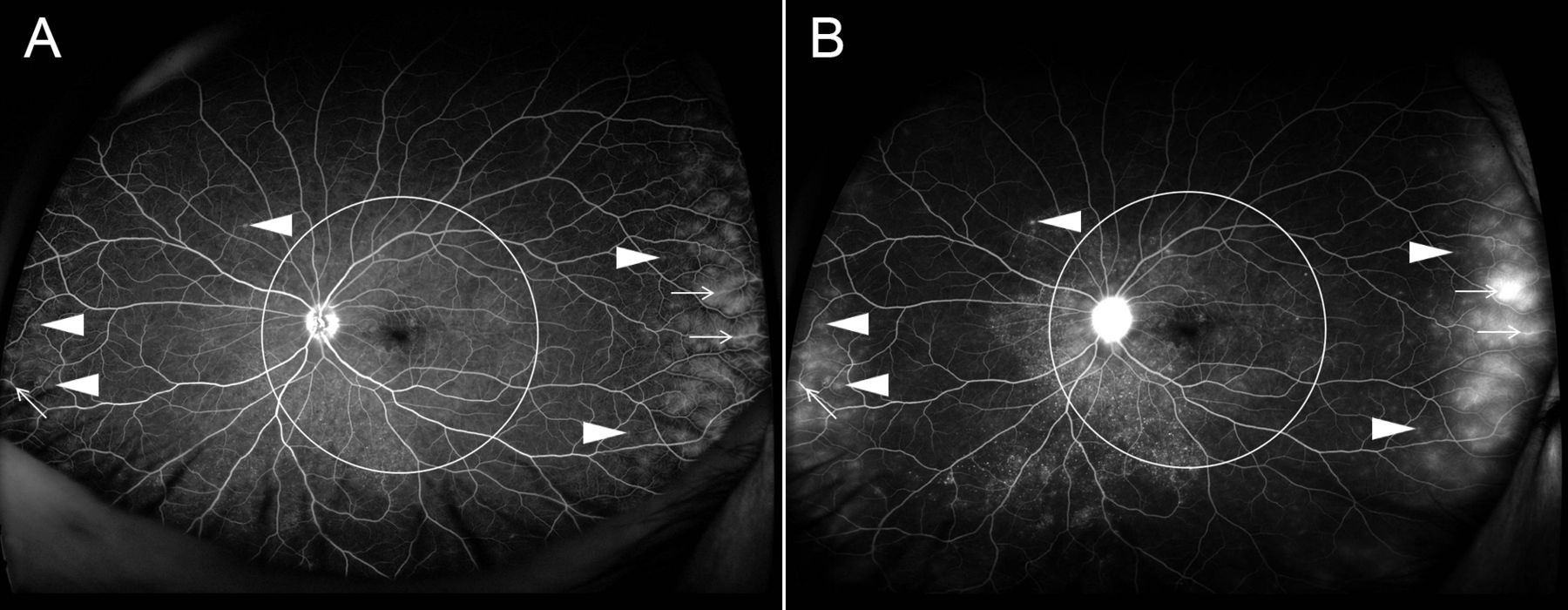
El síndrome de VKH afecta a múltiples órganos, entre los que se incluyen el sistema ocular, el sistema nervioso, el auditivo y la piel. Etiológicamente es desconocido, aunque se cree que hay una reacción de autoinmunidad contra los melanocitos uveales y cutáneos. El diagnóstico es clínico cumpliendo una serie de criterios ya establecidos, aunque la angiografía fluoresceínica, la tomografía de coherencia óptica y la punción lumbar pueden ser de gran ayuda tanto para el diagnóstico como para valorar posibles complicaciones.
¿Alguna duda? Puedes contactarnos para iniciar el tratamiento.


